Birdshot
Birdshot chorioretinopathy (BCR) is a bilateral, posterior uveitis that accounts for 0.6–1.5% of patients seen for uveitis in large referral centres and 6–7.9% of patients with posterior uveitis.1
Birdshot
Birdshot chorioretinopathy (BCR) is a bilateral, posterior uveitis that accounts for 0.6–1.5% of patients seen for uveitis in large referral centres and 6–7.9% of patients with posterior uveitis.1
Birdshot primarily affects Caucasians between the ages of 15 and 79 years (mean reported age at onset of 53 years), with blurred vision and floaters.1 In the initial stage the visual acuity (VA) is fairly symmetric between both eyes. In their review of 34 papers (pooled data of 213 patients), Shah et al. found a VA at presentation of 20/40 or better in 61.8% of eyes, of 20/50 to 20/160 in 23.8% of eyes and of 20/200 or less in 14.4% of eyes.1
Ophthalmologic Features and Evolution
The international consensus conference in 2006 has provided useful standardised research criteria for the clinical diagnosis of birdshot chorioretinopathy.2
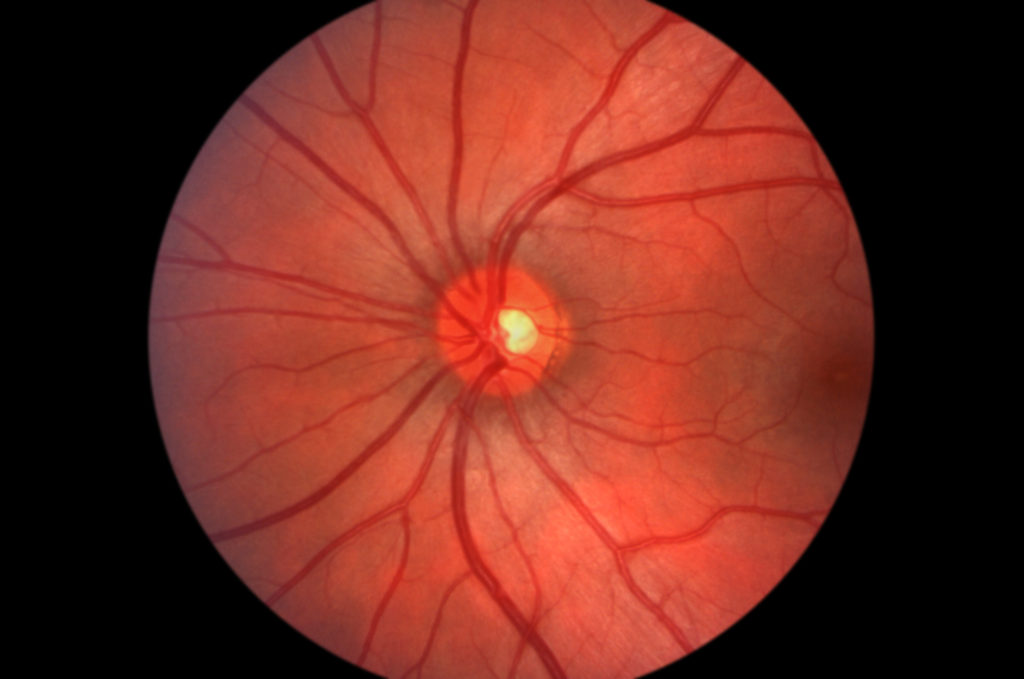
As in the original description of birdshot by Ryan and Maumenee in 1980, who compared the appearance of the fundus lesion to birdshot scattering from a shotgun, one typically observes bilateral oval or round (or irregular/linear in shape) hypopigmented choroidal spots in the posterior pole and midperiphery. The lesions measure about one-quarter to one-half disc diameter in size (see Figure 1a).
Lesions are localised around the optic disc and arcades, although they may be more numerous in the nasal fundus. In early disease it is not unusual to see asymmetrical ocular findings. Anterior segment is always mild (≤1+ cells, as defined by the international consensus conference),2 and posterior synechiae and keratic precipitates are absent.
Apart from the birdshot lesions, low-grade vitreous inflammation (≤2+ vitreous haze, as defined by the international consensus conference,2 without snowballs or snowbanking), optic disk oedema and retinal vasculitis (phlebitis in the posterior pole) are usually present. Monnet et al. observed macular oedema (determined by optical coherence tomography (OCT) criteria) in 34% of subjects with birdshot chorioretinopathy and have reported it as being the most common cause of decreased VA.1,3–5 Epiretinal membranes have been described in 19% of patients4 and, less commonly, choroidal neovascular membranes (CNV).
Controversy exists concerning visual outcome of patients with BCR. After a median follow-up of 3.5 years, Shah et al. showed a decline in VA of two or more lines of Snellen VA in 19.6% of eyes,1 with up to 45% of patients maintaining a VA of ≥20/40 in both eyes. Kiss et al. showed a preservation of visual function as determined by Snellen VAs and electroretinograms (ERGs) after treatment with immunosuppressive therapy. Approximately 80% of patients maintained or improved VA after a mean period of 81.2 months.6 Conversely, the worst outcomes were reported at 10 years of follow-up by Rothova et al.: VA was less than 20/200 in 39% of studied eyes despite the use of the recommended therapy.5
Fluorescein Angiography and Indocyanine Green Angiography
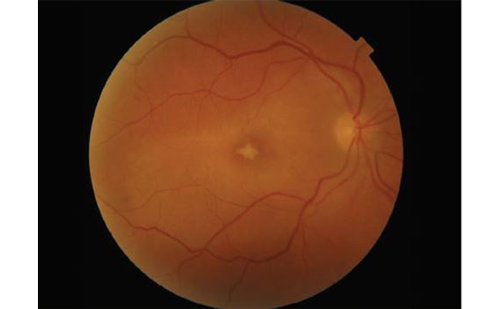
Fluorescein angiography (FA) usually shows hypofluorescent lesions in the early phase: in advanced disease, the lesions often become hyperfluorescent at the last phase. FA is most useful in demonstrating leakage from retinal vessels and disc hyperfluorescence, which is often subclinical but universally present (see Figure 1b). FA may also demonstrate cystoid macular oedema and CNV.
Indocyanine green angiography (ICG) is more useful than FA in demonstrating the birdshot lesions, which appear greater in number than clinically or on FA. These hypofluorescent, round–oval lesions are most visible during the intermediate phase of the angiogram and remain hypofluorescent in the last phase (see Figure 1c).7
Diagnosis
BCR is primarily diagnosed on clinical grounds, and other than human leukocyte antigen (HLA) A29, systemic investigations are normal.
The positive association of BCR with HLA A29 is well known, as 95.7% of cases are HLA A29-positive,1 preponderantly with one of the two main subtypes of HLA-A29.2. HLA A29 testing is thus one of the most sensitive tests in uveitis. A negative HLA A29 test should alert the clinician to doubt the diagnosis of BCR and search for alternative diagnoses, as fewer than 5% of the BCR cases reported in the literature are HLA A29-negative.
VA and inflammatory activity are poor makers for disease activity4 and we rely on additional tests such as visual fields and ERG testing to monitor disease severity.
Visual fields (automated perimetry 30-2) or Goldmann perimetry may show abnormalities in the majority of patients;8,9 a recent study has shown multiple foci as being the most common abnormality at baseline examination (27% of eyes), followed by arcuate (9% of eyes), enlarged blind spot (6% of eyes), central scotoma (5% of eyes) and undefined abnormality (38% of eyes).10 Furthermore, in the same study the mean deviation in authomated perimetry has been suggested as being the most appropriate monitoring parameter for BRC.
Electrophysiological investigations show delayed photopic 30Hz cone-derived flicker time, which represents the most sensitive marker of activity,11,12 maximal ERGs that are electronegative for most of the patients or a low a:b ratio.12 These findings reflect a primarily inner retinal dysfunction of cone and rod systems.8,12–14
Interestingly, Sobrin et al. have also suggested the use of rod, combined rod–cone, cone amplitudes, cone b-wave flash and flicker implicit times for detecting deterioration during observed intervals.15 Furthermore, the tapering of immunosuppressive agents could be initiated regarding the results of scotopic bright flash (combined rod–cone) amplitudes and photopic 30Hz flicker (cone) implicit times.11 The pattern ERG improvements possibly reflect the resolution of macular oedema.12
Treatments
Systemic corticosteroid (usually Prednisolone 1mg/kg per day to taper slowly followed by a maintenance dose) is the recommended therapeutic regimen. The majority of patients will require an immunomodulatory drug (ciclosporin,16 azathioprine,6,17 mycophenolate mofetil or methotrexate) as a steroid-sparing agent. Small studies have shown the efficacy of daclizumab (anti-interleukin 2 (IL-2) receptor antibody)15,18 and anti-tumour necrosis factor-alpha (TNF-γ) therapies in BCR.19
Differential Diagnoses
Sarcoidosis is probably the disease that can most easily mimic birdshot lesions due to the appearance of choroidal granulomas. Tuberculosis (TB) usually has other ocular or systemic features to distinguish it. Syphilis and Lyme disease are great masqueraders and may present with vitritis and choroidal lesions.
The other white dot syndromes can be easily confused with birdshot chorioretinitis. However, careful attention to clinical features, findings and natural history will usually distinguish BCR from the other syndromes.
Primary central nervous system (CNS) intraocular lymphoma may simulate birdshot lesions by presenting as a bilateral vitritis with sub-retinal/retinal pigment epithelium (RPE) infiltrates in the presence of a normal systemic work-up. The presence of yellowish extensive confluent sub-retinal lesions only partially responsive to steroids may alert the physician. Nevertheless, a high index of suspicion for this rare disease is needed.
Acute Posterior Multifocal Placoid Pigment Epitheliopathy
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) affects individuals between the ages of 11 and 66 years, with a mean age at onset of 26.5 years.20 Affected individuals report symptoms of blurred vision, scotomas and photopsias that follow a recent history of viral illness in one-third of patients.21 An association with HLA B7 and HLA DR2 has been described,22 but is probably too low to be useful in diagnosis.
Clinical
APMPPE is characterised by bilateral (simultaneous or sequentially within a few days), multifocal yellowish-white placoid lesions on fundoscopic examination (see Figure 2a). The lesions measure less than one disc diameter in size and are spread through the posterior pole at the level of RPE with a tendency to scar, leaving RPE atrophy and hyperpigmentation. Lesions may be confluent. Over a few weeks, one characteristically sees new active lesions develop, as older lesions become pigmented and inactive, within the same eye.
Various degrees of ocular inflammation (presence of cells in the anterior chamber or in the vitreous) are common findings.
The prognosis of visual function in APMPPE is good. The majority have spontaneous recovery of visual function to ≥20/40 within three–six weeks of the onset of the disease.23 A minority of patients have been reported as having a deterioration of function related to extensive RPE changes or choroidal neovascularisation.
Fluorescein Angiography and Indocyanine Green Angiography
Active lesions are hypofluorescent in the early stage (see Figure 2b) and become hyperfluorescent at a later stage (see Figure 2c). A prolonged filling of the choroid is also frequent.
ICG appearance of APMPPE is characteristic of choroidal non-perfusion with hypofluorescence of active and healed lesions.
Treatment
In most cases, no treatment is required as the natural history is good. We have occasionally seen a variant of APMPPE characterised by chronicity and extensive macular involvement with acuity loss. We treat such cases with tapering oral steroids. Recent case series have suggested that the visual prognosis is less favourable than that originally described by Gass. It is our current practice to treat any patient with macular lesions with systemic steroids, although the benefit of systemic steroids has not been demonstrated via randomised treatment control trials. In addition, there is a very rare association of APMPPE with CNV vasculitis.23 These patients would be treated with systemic steroids and/ or immunosuppression.
Differential Diagnoses – Other White Dot Syndromes
TB choroiditis can present a similar picture and should be considered in at-risk individuals and patients from areas of high TB prevalence. Tuberculous skin testing (Mantoux) or gamma interferon (IFN-γ) testing (QUANTIferon) may assist, but the diagnosis remains challenging as most patients have extra-pulmonary disease and therefore normal chest radiology.
Posterior placoid syphilitic choroidopathy can have a similar appearance. Unlike APMPPE, there is no early hypofluorescence of the lesions on angiography.
Multiple Evanescent White Dot Syndrome
First reported in 1984 by Jampol et al., multiple evanescent white dot syndrome (MEWDS) is a usually unilateral, idiopathic posterior uveitis characterised by discrete white lesions on fundus examination. A viral aetiology has been suggested, combined with an immune-mediated mechanism.24 Patients affected are usually young adults. Symptoms include blurred vision; photopsias, positive scotomas and enlargement of the blind spot.
Clinical
The MEWDS lesions of the fundus are grey-white, multiple, ranging from 100 to ≥200μm in size and localised in the inner retina, at the RPE level and inner choroid (see Figure 3a).25 The lesions appear in the posterior pole and extend to the midperiphery but are mostly scattered in the paramacular area.
A granular appearance of the macula, often present, is pathognomonic. Vitreous cells and mild inflammation of the optic nerve25 have also been described.
Fluorescein Angiography and Indocyanine Green Angiography
The lesions are characterised as wreath-like punctuate areas of hyperfluorescence on early phase on FA (see Figure 3b) with late staining.25 ICG is strikingly characteristic and shows multiple hypofluorescent lesions more numerous than those seen on clinical examination or FA (see Figure 3c).25
There is no need for further investigations other than clinical examination and FA since MEWDS resolves spontaneously without any treatment with a return to the baseline vision in about six to eight weeks.
Differential Diagnoses – Other White Dot Syndromes
The authors have seen syphilitic posterior uveitis occasionally present with multiple white chorio-retinal lesions than can mimic MEWDS, especially with co-existing human immunodeficiency virus (HIV) disease.
Multifocal Choroiditis
Multifocal choroiditis (MFC) is a presumed autoimmune inflammatory disease primarily affecting myopic young women in their third to fifth decades.
Fundus examination shows small, round chorioretinal yellowish lesions (50–350μm in size) (see Figure 4a) that become atrophic in later stages. The distribution of the scars is variable: scattered or in aggregates widespread in the posterior pole and/or the periphery.
Clinically, vitreous cells can be present in active disease. Complications include sub-retinal neovascular membranes in one-third of patients.26
Fluorescein Angiography and Indocyanine Green Angiography
In the acute phase, there is early hypofluorescence on FA and late hyperfluorescence (see Figure 4b) in areas corresponding to hypofluorescent dots on ICG (see Figure 4c). On late phase, ICG shows window defects and masking effects corresponding to chorioretinal scars. ICG identifies hypofluorescent spots within the choroid, often more numerous than on ophthalmoscopy.
Evolution
MFC follows a chronic relapsing course.
Treatment
Systemic and local steroids, combined with immunosuppressive drug therapy, should be considered.
Choroidal neovascular membranes can be treated with photodynamic therapy,27,28 high-dose steroids and more recently, with anti-vascular endothelial growth factor (VEGF) therapy.29
Differential Diagnosis
Sarcoidosis and Tuberculosis
Presumed ocular histoplasmosis syndrome: similarities with multifocal punched-out chorioretinal scars and CNV, but absence of intraocular inflammation and fewer lesions (five or fewer) in endemic regions. Punctate inner choroidopathy (PIC) is characterised by similar multifocal choroiditis lesions and CNV, except that there is an absence of intraocular inflammation and that the disease is preponderant among younger myopic females.
Punctate Inner Choroidopathy
PIC was first described by Watzke et al. in 1984. PIC predominantly affects young women between the ages of 16 and 40. Most patients are myopic and the disease may present initially unilaterally. Symptoms include sudden onset of blurred vision, central or paracentral scotomas or photopsias.
The anterior segment and the vitreous appear quiet. Retinal examination shows one or more characteristic PIC lesions that are initially yellow, measure 100–300μm in diameter and are located at the level of the RPE and the inner choroid. PIC patients typically have numerous spots confined to the posterior pole and also in the midperiphery. The major complication is the development of sub-retinal neovascular membranes (see Figures 5a and 5b) in 25% of PIC eyes. Haemorrhage and scarring of foveal or juxtafoveal membranes will result in severe acuity loss if untreated.
Fluorescein Angiography
Hyperfluorescence of the dots (early phase) and late phase leakage. Leakage of CNV membranes in the early phase.
Treatment
It is our practice to treat acute macular PIC lesions with systemic steroids to prevent the development of neovascular membranes. For established membranes, anti-VEGF intravitreal injections are the treatment of choice for foveal and juxtafoveal membranes. More destructive treatments such as argon laser and photodynamic therapy remain options for extrafoveal membranes.30,31
Differential Diagnosis
Sarcoidosis, presumed ocular histoplasmosis.
Serpiginous Choroiditis
Serpiginous choroiditis (SC) is a rare, bilateral, chronic, recurrent inflammatory disease affecting the choroid and the RPE. Healthy young to middle-aged men (30–60 years old) are mainly affected. HLA B7 has been found to be prevalent in patients with SC.32 The pathogenesis is unknown but an infectious aetiology is suspected, possibly primary bacterial or secondary to an immunological process. TB has been postulated.33
Patients usually experience blurred vision, photopsias, scotomas, metamorphopsias and visual field loss. The anterior segment is quiet although a mild vitritis is observed. On fundus examination, the active lesions appear geographic in shape, creamy yellow sub-retinal infiltrates localised originally in the peripapillary area and progressing centrifugally. Based on clinical presentation, Lim et al. have classified SC into peripapillary, macular and ampiginous types.34 In the convalescent phase, the lesions leave atrophic pigmented scars.
Fluorescein Angiography and Indocyanine Green Angiography
FA shows early hypofluorescence and late hyperfluorescence in the border of active lesions. ICG is the investigation of choice to establish the severity of choroidal involvement.34 ICG of acute lesions shows geographic hypofluorescence with perilesional hyperfluorescence. Hypofluorescence on ICG indicates chorioretinal scarring. Recently, fundus autofluorescence has been shown to be useful to detect RPE changes in acute episodes of SC.35
Steroid and immunosuppressive drug therapies (ciclosporin A, azathioprine or mycophenolate mofetil) have been found to be effective in treating this disease.
Differential Diagnosis
Choroidal TB, APMPPE, multifocal choroiditis, syphilis and outer retinal toxoplasmosis are the differential diagnoses.
Conclusion
Table 1 summarises clinical and angiographic findings and differential diagnoses of white dot syndromes. ■