Retinitis pigmentosa (RP) is an inherited retinal dystrophy characterised with the progressive loss of photoreceptors.1,2 Night blindness together with the peripheral visual field loss is the most prominent clinical feature.1,2 Central vision is relatively spared up to the later stages of the disease process.3 However, when macular complications such as macular oedema arise, central vision is often dramatically impaired even in the early stages of the disease. The prevalence of cystoid macular oedema (CME) is reported to be 10–20% in the eyes of patients with RP.4,5
The pathogenesis of RP-related CME is still unclear. Blood–retinal barrier (BRB) impairment, retinal pigment epithelium (RPE) pump disturbance, inflammation, autoimmunity and vitreoretinal interface changes may be among the potential causes of CME.6–19
Fishman et al.6 examined 15 RP patients with vitreous fluorophotometry. All patients showed abnormally high concentrations of fluorescein within the vitreous which was a sign of BRB abnormality. Larsen et al.7 demonstrated that BRB leakage was markedly increased in six RP patients with the help of ocular spectrofluorophotometry. Vinores et al.9 investigated the immunohistochemical staining for albumin on paraffin sections of 22 normal and 29 RP eyes. Electron microscopic immunocytochemical staining for albumin was performed on additional six normal and nine RP-affected eyes. Two-thirds of the eyes with RP demonstrated extravascular albumin in the inner portion of the posterior retina. This was evident even in the absence of CME but eyes with CME showed extensive BRB failure. Küchle et al.10 analysed the BRB in patients with RP with a laser cell photometer in 56 eyes of 29 patients and found that aqueous flare values were higher in patients with RP than in controls; moreover, patients with RP-related CME had even higher values with a mean of 14.66 photon counts per millisecond compared with 9.65 for patients with RP but having no CME. RPE seems to lose polarised apical distribution in association with macular oedema and RP and cannot effectively pump out ions and fluid from the outer retina.11
Autoimmune processes and/or inflammation can be deemed as the hallmark of RP-related CME. Spalton et al.12 examined 25 patients with RP and all patients had an excessive number of vitreous cells; six even had exudates in the pre-equatorial fundus indistinguishable from pars planitis. They suggested that this might be a general response noticed in many types of tapetoretinal degeneration in reaction to actually degenerating photoreceptors or RPE. Furthermore, Heckenlively et al.13 looked for the presence of antiretinal antibodies in a group of 30 consecutive patients with CME and RP, 30 consecutive patients with RP but without CME and 50 normal subjects. Twenty-seven (90%) of the patients with RP and CME had antiretinal protein antibody activity compared with three of 50 normal subjects (6%) and only four of 30 patients (13%) with RP but without coexistent CME.
Yoshida et al.14 studied the nature of inflammatory reaction in eyes of patients with RP. In 190 of 509 eyes (37.3%) with RP, ‘1+’ (5–9 cells per field) or more cells were observed in the


anterior vitreous cavity. A strong inflammatory reaction with ‘2+’ cells (10–30 cells per field) was associated with younger age. The levels of various proinflammatory cytokines and chemokines including monocyte chemotactic protein-1 were increased both in the aqueous humour and vitreous fluid of RP patients compared with the levels in control patients.14
Vitreoretinal abnormalities can be a factor in the development of CME in RP patients.15–17 Kim et al.16 detected CME in 46 of 220 eyes with RP with the spectral domain optic coherence tomography (SD-OCT). Vitreomacular interface abnormalities including epiretinal membrane development and vitreomacular traction were evident in over 40% of patients and the presence of vitreoretinal abnormalities was significantly higher in eyes with CME than in eyes without CME (64.1% versus 36.8%). Spontaneous release of vitreoretinal traction may alleviate the RP-related CME as soon as the spontaneous resolution of traction occurs.17 It is also speculated that vitreoretinal abnormalities may induce a mechanical damage to the Muller cells that are an essential element for visual transduction and retinal homeostasis as Muller cells play an important role in the fluid dynamics.18,19
Treatment
Various treatment alternatives including topical (nonsteroidal antiinflammatory agents, carbonic anhydrase inhibitors), oral agents (carbonic anhydrase inhibitors, steroids), grid laser photocoagulation and vitreoretinal surgery were utilised in the treatment of RP-related CME.11,20–25 In this review we focus on the intravitreal pharmacotherapy.

Intravitreal steroids
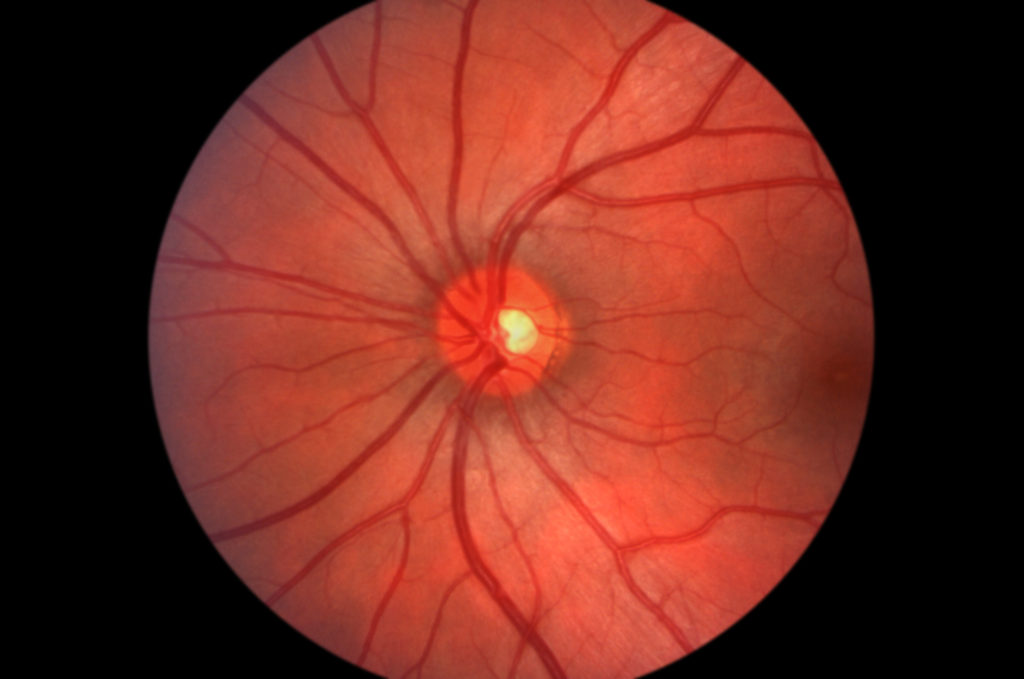
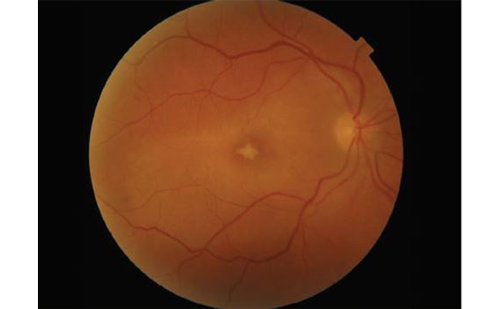
Steroids exert their effect via several mechanisms such as reduction in synthesis and release of proinflammatory cytokines, suppression of the production and migration of inflammatory cells, and suppression of the autoimmune process and restoration of the integrity of BRB.26–30 The previous reports on intravitreal steroid administration in eyes with RP-related CME30–39 are summarised in Table 1. The treatment effect of dexamethasone implant can be seen in a case with bilateral RP-related CME (Figures 1 and 2).
The most important drawbacks in the use of intravitreal steroids in RP-related CME are temporary drug effect and risk of ocular hypertension and cataract formation especially with consecutive injections.30
Intravitreal anti-vascular endothelial growth factor agents
Intravitreal anti-vascular endothelial growth factor (VEGF) administration is shown to be very effective in VEGF-driven disease processes by reversing mainly the VEGF-driven BRB disruption.40,41
Salom et al.42 quantified the VEGF-A levels in aqueous humour of 16 eyes of 16 patients with RP. They found that aqueous VEGF-A levels were markedly lower in eyes with RP than in control patients. Strong et al.43 hypothesised that a localised VEGF production under pathologic conditions, for example by Muller cells, may contribute to CME formation in RP patients and this fact may also be the reason for the rarity of peripheral neovascularisation in eyes with RP. The previous reports on intravitreal anti-VEGF agent administered in eyes with RP-related CME43–49 are summarised in Table 2.
Intravitreal autologous bone marrow-derived heamatopoietic stem cell transplantation
Siqueira et al.50 published a case with RP-related CME who received treatment with an intravitreal injection of autologous bone marrowderived heamatopoietic stem cell (auto-BMHSCT) and showed complete

resolution of CME 7 days after the injection. This effect lasted for a month. Visual acuity was improved to 20/50 from 20/32. Macular sensitivity measured by the microperimetry was also improved. Authors speculated that auto-BMHSCT might restore the BRB via its paracrine effects and thereby provided the resolution of the oedema.
Comments There is yet no proven treatment for RP-related CME and no randomised controlled studies with a sufficient number of patients have been performed. Intravitreal steroids and anti-VEGF agents are reported to have some effect on RP-related CME but the durability of treatment effect and the selection of agent (anti-VEGF or steroids) are highly controversial. Thereby, we intend to review the current status of the intravitreal pharmacotherapy in the treatment of RP-related CME and share some of the latest data with the ophthalmic community.