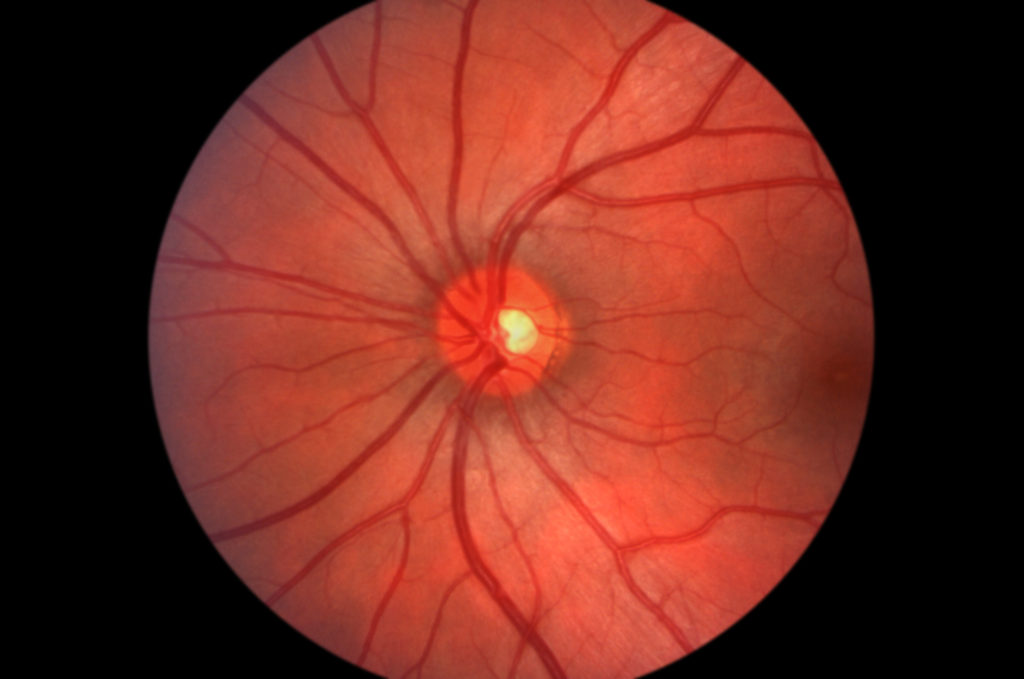
Retinitis pigmentosa (RP) is a group of disorders whose main clinical features include diffuse photoreceptor dysfunction, diminished electroretinogram (ERG), and progressive visual field loss. The disorders affect approximately one in 5000 people worldwide.1 Clinical features often include peripheral retinal pigment epithelial atrophy, ‘bone spicule’ pigment clumping, waxy optic disk pallor and arteriolar attenuation. Patients often experience nyctalopia as an early symptom followed by progressive visual field loss and eventual central vision loss. Pigmented vitreous cells, posterior subcapsular cataract, and cystoid macular edema have also been associated with RP.
Interestingly, RP has been associated with all forms of mendelian inheritance patterns. It appears that autosomal dominant is the most common identifiable inheritance pattern, present in approximately 15–25% of cases. Autosomal recessive inheritance is also common, as is the absence of any family history. X-linked and mitochondrial variants have also been described. There are over 100 genes that have been identified and implicated in the disease.1,2 These defects are organized by chromosome in the RetNet subdivision of the Online Mendelian Inheritance in Man database (OMIM™). Many mutations, including the first gene discovered to cause RP, are defects in rhodopsin, the pan-rod photopigment. Many novel genetic defects in products involved in the visual pigment transduction pathway continue to be described. Among these are rhodopsin (RHO), RDS/peripherin, pre-messenger RNA (mRNA) processing factor 31 (PRPF31), RP31, Prominin 1 (PROM1), RP GTPase regulator (RPGR) and RP25. In addition, there is a wide range of disease severity and phenotypic expressivity linked to specific mutations. This is especially evident in analysis of the RDS/peripherin gene2 located on chromosome 6p whose product localizes to a protein present in the peripheral outer rod segment. Various mutations may occur with a wide range of phenotypes, ranging from pattern dystrophies to Stargardt’s disease to RP, even within the same family.
There has been no proven effective treatment for RP. Observations that patients taking vitamin A may have a slower decline in ERG and photoreceptor function led to a randomized controlled trial investigating vitamin A and E supplementation.3,4 The results demonstrated a role for vitamin A supplementation in retarding ERG amplitude loss but no obvious significant effect on vision and visual field loss. A subsequent analysis of patients deemed to have more accurate visual field measurement demonstrated slower rates of visual field loss in those using vitamin A.5,6 Larger trials with longer follow-up are needed to completely assess the efficacy of this treatment. However, many retinal specialists recommend 15,000IU of vitamin A palmitate in patients with RP excluding women who may be pregnant or are planning to become pregnant.
The future of RP treatment will rest upon pharmacogenetic and molecular biologic techniques for replacing defective genes, restoring the function of defective proteins, and preventing abnormal photoreceptor function; with the use of implantable electrical devices in advanced cases. Gene therapy has undergone many recent advances, particularly involving delivery of functional genes through viral vectors, especially the adeno-associated viral vector (AAVV),7,8 which has been shown to efficiently transmit DNA to both photoreceptor and retinal pigment epithelium (RPE) cells.7–9 Several essential characteristics of this therapy must be considered in order to evaluate therapeutic feasibility.10 They include: must be a monogenic loss-of-function mutation (which comprises only a small fraction of RP pathology), the specific genetic defect must be identifiable, the transgene vector must have low immunogenicity, and the expression of the transgene must occur long term. Despite these obstacles, significant progress has been made in this area. In mice, AAVV delivery via subretinal injection has been used to prevent expression of non-functional rhodopsin and deliver functional rhodopsin, resulting in improved ERG response.11 Another strategy that has been investigated is the use of vectors to photosensitize cells in the retina other than photoreceptors. Intravitreal injection of AAVV in rats has shown to be capable of inducing expression of an additional opsin, channelopsin 2 (CHOP2), in retinal ganglion cells, leading to an improved ERG response. As the vast array of genetic defects and expressions of RP are elucidated along with advancements in molecular biologic techniques, gene delivery and protein–protein interaction will no doubt become a major player in the future of treatment.
Furthermore, significant progress is being made in the field of retinal prosthesis.12–16 These devices include some method of transmitting electrical signals to remaining inner retinal cells in patients with damaged photoreceptors. The feasibility of this technology was determined by showing that significant proportions of the inner retinal elements, namely ganglion cells, remain histologically intact even in patients with significant photoreceptor loss from RP.14 The first US clincal trial was performed by Optobionics with an artificial silicone retina consisting of a subretinal microphotodiode array.15 The device was found to be safe with many patients developing evidence of improved visual function. Currently, the most advanced device employs an epiretinal microarray sponsored by Second Sight Medical Products and is currently in phase II clinical trials. This employs a tiny camera mounted on the patient’s eyeglasses that transmits a digital image to the intraocular microarray, which in turn transmits the image to electric impulses that can be detected by the retina (see Figure 1).16
Leber Congenital Amaurosis
Leber’s congenital amaurosis (LCA) is a group of visual disorders characterized by infantile onset, recessive inheritance, and severe visual loss secondary to severe rod–cone dysfunction.17,18 Classically these diseases have been classified as infantile-onset RP. The incidence is rare, occuring in between one in 30,000 and one in 80,000 live births. Affected children develop severe visual loss and wandering nystagmus, and often demonstrate the oculo-digital reflex. Patients typically have amaurotic pupils and absent ERG response.19 Early in the disease course, the fundus often appears normal. Many patients eventually develop pigmentary retinopathy and, rarely, bone spicule formation. Cataract and keratoconnus may be associated. At least 14 gene mutations have been identified and are thought to account for approximately 70% of cases.20–23 These genes are involved in phototransduction, structural proteins, the vitamin A cycle, and outer photoreceptor segment phagocytosis. Affected patients typically have visual acuity ranging from 20/200 to no light perception. Visual acuity usually remains stationary or shows slow progressive decline.
Although the disorder is heterogenous in terms of underlying defect, molecular diagnosis is important for genetic counseling and possible treatment.21,24 In the pre-genomic era, there were no effective treatments for the disorder. Recently, two clinical trials have investigated the use of a recombinant AAVV-delivered coding sequence of RPE65, a gene expressed by the RPE and necessary for the visual pigment cycle.25,26 RPE65 is normally required for the regeneration of 11-cis retinal for phototransduction. LCA2 is caused by mutations in the 65Da gene product, which is an RPE-specific protein. Initial evidence for the use of this therapy has come about through observations that while photoreceptors are dysfunctional from birth, photoreceptor cell death occurs later in the disease process. In addition, a naturally occuring animal model (Swedish Briard Dog) has been used to show improved visual function with subretinal delivery of AAVV RPE65.23–27 Interestingly, this method of delivery, using complementary DNA (cDNA) delivered via viral vector, has been shown to cause sustained expression of the gene product in animal models. Bainbridge et al. investigated three young adult patients by administering subretinal AAVV RPE65.28 Of these, one patient developed evidence of visual improvement measured by microperimetry and dark adapted perimetry as well as subjective tests of visual mobility. There were no severe untoward complications of transvitreal/subretinal delivery of the vector. There were, however, no significant visual changes in acuity or Goldmann perimetry. Maguire et al. performed a similar trial with subretinal delivery of AAVV RPE65 to three young adults with LCA2.29 All patients developed improved pupillary responses, decreased amplitude of nystagmus, and improved subjective visual acuity. One patient developed a macular hole that was thought to be related to a pre-existing epiretinal membrane that was not removed prior to surgery. Both of these trials, although unmasked and non-placebo-controlled, represent steps forward in treatment in a previously untreatable blinding retinal disorder. With longer follow-up, larger trials and improved techniques the efficacy of this treatment will continue to be elucidated. As described above, retinal prostheses may also be a feasible option in the near future for patients with LCA.
X-linked Retinoschisis
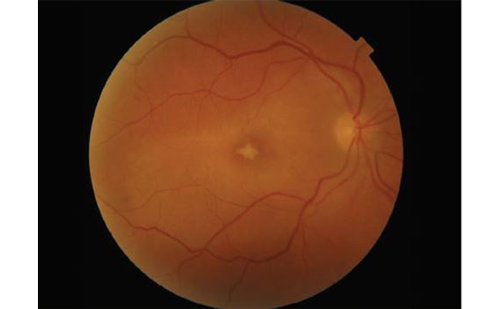
X-linked retinoschisis, or inherited splitting of the foveal nerve fiber layer, is the leading cause of macular degeneration in young males, with a prevalence between one in 15,000 and one in 30,000 males.30,31 The disorder follows typical X-linked inheritance with no male–male transmission, carrier females, and affected males.32 The disorder is caused by several identified mutations in retinoschisin, a retinal protein product of the gene RS1.33–35 RS1 maps to the distal arm of the X-chromosome (Xp22). Retinoschisin is a secreted protein that likely participates in cell–cell interaction. Genetic defects cause failure of secretion or failure of extracellular polymerization and cell adhesion. Clinically, the disorder has a heterogenous set of features. There appears to be a bimodal distribution of age at diagnosis.36 Patients may present in infancy with squinting and nystagmus with severe vision loss or at school age with decreased visual acuity. Interestingly, there appears to be no correlation with specific genetic mutation and disease severity, even within affected families. Visual acuity can range from 20/20 to severe visual loss.37,38 Physical exam discloses a wheel-like pattern of foveal retinal splitting often extending to the retinal periphery. These findings may be dramatic in childhood but typically become less obvious with time as schisis cavities regress, often leaving a pattern of hyperpigmentation over time. Diagnosis is suspected with typical fundus features as well as the presence of diminished B-wave amplitude on ERG with relatively preserved A wave, reflecting damage to inner retinal elements.
The disease course may be variable, but typically demonstrates variable visual loss that is stable initially, with eventual decline beginning in the fourth to fifth decade of life.30,31,37,38 Furthermore, additional retinal complications such as macular traction/detachment, vitreous hemorrhage, and peripheral retinal breaks may occur.39 Genetic testing is available and is an important component of diagnosis and genetic counseling for patients and their families. Low vision aids and appropriate refraction are major elements for supportive treatment. A recent report described the use of topical dorzolamide, which appears useful in decreasing cystic retinal spaces and gives possible improvement in vision.40 The usefulness of this treatment modality requires further investigation and follow-up.
Genetic therapy for X-linked retinoschisis may be feasible, but will have some unique challenges. Mouse models of the disorder exist and recently have been used to attempt gene-delivery-based rescue.41,42 The use of AAVV gene replacement has been shown to be capable of improving B-wave ERG. This treatment modality may have some role in treatment, but will likely apply to eyes with early diagnosis with preserved retinal structural elements. Time and experience will tell whether this animal-modeled treatment may extrapolate to human disease.
Stargardt’s Disease
Stargardt’s disease, or fundus flavimaculatus, is an autosomal recessively inherited form of macular degeneration and is the most common macular dystrophy in patients under 50 years of age.43 The disorder affects approximately one in 10,000 individuals. The most common genetic defect is in ABCA4 on chromosome 1p, whose product is the ATP-binding cassette transporter protein.44 The protein localizes to rod outer segments. Stargardt’s disease can be highly variable in terms of clinical features. As with several other genes implicated in hereditary retinal dystrophies, patients with deficiencies in ABCA4 can display Stargardt’s phenotype, an RP-like picture, or pattern dystrophy even within the same family.44–47 Several mutations have and continue to be identified, and there is some evidence that different mutations may be associated with distinct phenotypes and disease severity.44–46 Patients are typically young and present with gradual decline in visual acuity ranging from mild impairment to visual acuity of 20/200. Physical examination reveals loss of foveolar reflex, deep yellow ‘pisciform’ subretinal deposits, and pathognomonic dark choroid on fluorescein angiogram.46–48 The angiographic finding is related to diffuse blockage by excess RPE lipofuscin accumulation.46 Visual field testing is often normal early in the disease course but progresses to central scotoma.47 ERG is typically normal early in the disease but may progress to prolonged implicit times or generalized depression of amplitude late in the disease course.48 Histopathologic studies support the notion that the underlying genetic defect leads to the accumulation of lipofuscin,49 a metabolic waste product and subsequent retinal and RPE dysfunction as a result of photoreceptor cell loss.49–51 Although genetic advances are being made in understanding Stargardt’s disease, the molecular mechanism by which the disease manifests are not well understood. Currently there is no proven treatment; further research will be needed to investigate the feasibility of gene therapy for Stargardt’s disease.
The classification, diagnosis, and now treatment of hereditary retinal degeneration have changed significantly in our era of molecular biology and genetic evaluation. The genetic testing allows for accurate diagnosis, while providing information for counseling for the patient and the family. Advances in molecular biology have provided potential treatment through gene and drug therapy. ■